Sequence alignment revealed two regions of similarity between the FT and Col4 (red boxes). Yellow and green shades represent high and moderate similarity, respectively. ( Dotted line, non-homologous residues; asterisks, identical residues. )
Figure from: Küffer A Lakkaraju AK, Mogha A. et al. Nature. 2016 Aug 8. doi: 10.1038/nature19312
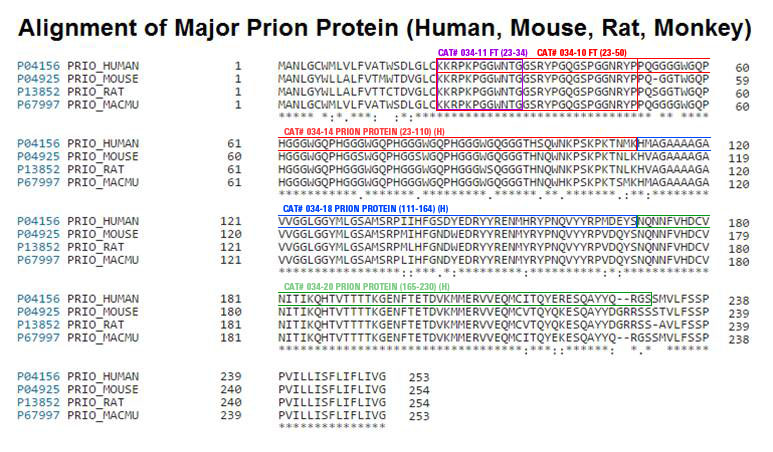
Ablation of the cellular prion protein PrPC leads to a chronic demyelinating polyneuropathy affecting Schwann cells. Neuron-restricted expression of PrPC prevents the disease, suggesting that PrPC acts in trans through an unidentified Schwann cell receptor. Here we show that the cAMP concentration in sciatic nerves from PrPC-deficient mice is reduced, suggesting that PrPC acts via a G protein-coupled receptor (GPCR). The amino-terminal flexible tail (residues 23-120) of PrPC triggered a concentration-dependent increase in cAMP in primary Schwann cells, in the Schwann cell line SW10, and in HEK293T cells overexpressing the GPCR Adgrg6 (also known as Gpr126). By contrast, naive HEK293T cells and HEK293T cells expressing several other GPCRs did not react to the flexible tail, and ablation of Gpr126 from SW10 cells abolished the flexible tail-induced cAMP response. The flexible tail contains a polycationic cluster (KKRPKPG) similar to the GPRGKPG motif of the Gpr126 agonist type-IV collagen. A KKRPKPG-containing PrPC-derived peptide (FT23-50) sufficed to induce a Gpr126-dependent cAMP response in cells and mice, and improved myelination in hypomorphic gpr126 mutant zebrafish (Danio rerio). Substitution of the cationic residues with alanines abolished the biological activity of both FT23-50 and the equivalent type-IV collagen peptide. We conclude that PrPC promotes myelin homeostasis through flexible tail-mediated Gpr126 agonism. As well as clarifying the physiological role of PrPC, these observations are relevant to the pathogenesis of demyelinating polyneuropathies-common debilitating diseases for which there are limited therapeutic options.
K¨¹ffer A Lakkaraju AK, Mogha A. et al. Nature. 2016 Aug 8. doi: 10.1038/nature19312
Mammalian prions, transmissible agents causing lethal neurodegenerative diseases, are composed of assemblies of misfolded cellular prionprotein (PrP). A novel PrP variant, G127V, was under positive evolutionary selection during the epidemic of kuru--an acquired prion disease epidemic of the Fore population in Papua New Guinea--and appeared to provide strong protection against disease in the heterozygous state. Here we have investigated the protective role of this variant and its interaction with the common, worldwide M129V PrP polymorphism. V127 was seen exclusively on a M129 PRNP allele. We demonstrate that transgenic mice expressing both variant and wild-type human PrP are completelyresistant to both kuru and classical Creutzfeldt-Jakob disease (CJD) prions (which are closely similar) but can be infected with variant CJD prions, a human prion strain resulting from exposure to bovine spongiform encephalopathy prions to which the Fore were not exposed. Notably, mice expressing only PrP V127 were completely resistant to all prion strains, demonstrating a different molecular mechanism to M129V, which provides its relative protection against classical CJD and kuru in the heterozygous state. Indeed, this single amino acid substitution (G→V) at a residue invariant in vertebrate evolution is as protective as deletion of the protein. Further study in transgenic mice expressing different ratios of variantand wild-type PrP indicates that not only is PrP V127 completely refractory to prion conversion but acts as a potent dose-dependent inhibitor of wild-type prion propagation.
Asante EA, Smidak M, Grimshaw A et al., Nature. 2015 Jun 25;522(7557):478-81. doi: 10.1038/nature14510. Epub 2015 Jun 10.
GPR126 is an orphan heterotrimeric guanine nucleotide-binding protein (G protein)-coupled receptor (GPCR) that is essential for the development of diverse organs. We found that type IV collagen, a major constituent of the basement membrane, binds to Gpr126 and activates its signaling function. Type IV collagen stimulated the production of cyclic adenosine monophosphate in rodent Schwann cells, which require Gpr126 activity to differentiate, and in human embryonic kidney (HEK) 293 cells expressing exogenous Gpr126. Type IV collagen specifically bound to the extracellular amino-terminal region of Gpr126 containing the CUB (complement, Uegf, Bmp1) and pentraxin domains. Gpr126 derivatives lacking the entire amino-terminal region were constitutively active, suggesting that this region inhibits signaling and that ligand binding relieves this inhibition to stimulate receptor activity. A new zebrafish mutation that truncates Gpr126 after the CUB and pentraxin domains disrupted development of peripheral nerves and the inner ear. Thus, our findings identify type IV collagen as an activating ligand for GPR126, define its mechanism of activation, and highlight a previously unrecognized signaling function of type IV collagen in basement membranes.
Paavola KJ, Sidik H, Zuchero JB et al., Sci Signal. 2014 Aug 12;7(338):ra76. doi: 10.1126/scisignal.2005347.