Table modified from: Steward AF. Diabetes. 2014 Apr;63(4):1198-9.
| Caerulein |
069-01 |
1352.55 |
pGlu-Gln-Asp-Tyr(SO3 H)-Thr-Gly-Trp-Met-Asp-Phe-NH2 |
CCK(26-33) |
069-03 |
1143 |
Asp-Tyr(SO3H)-Met-Gly-Trp-Met-Asp-Phe-NH2 |
CCK(27-33) |
069-05 |
947 |
Tyr-Met-Gly-Trp-Met-Asp-Phe-NH2 |
Gastrin-related peptide |
027-09 |
710.88 |
Aoc-Trp-Met-Asp-Phe-NH2 |
Pentagastrin |
027-21 |
767.38 |
tBoc -β-Ala-Trp-Met-Asp-Phe-NH2 |
Gastrin (4-17)/ minigastrin (Human) |
027-20 |
1831.72 |
Trp-Leu-Glu-Glu-Glu-Glu-Glu-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2 |
Gastrin I (Human) |
027-04 |
2098.23 |
pGlu-Gly-Pro-Trp-Leu-Glu-Glu-Glu-Glu-Glu-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2 |
Xenin (18-25) / Xenin-8 (Human) |
046-95 |
1046.28 |
His-Pro-Lys-Arg-Pro-Trp-Ile-Leu |
Mammalian cells secrete a large number of small proteins, but their mode of translocation into the endoplasmic reticulum is not fully understood. Cotranslational translocation was expected to be inefficient due to the small time window for signal sequence recognition by the signal recognition particle (SRP). Impairing the SRP pathway and reducing cellular levels of the translocon component Sec62 by RNA interference, we found an alternate, Sec62-dependent translocation path in mammalian cells required for the efficient translocation of small proteins with N-terminal signal sequences. The Sec62-dependent translocation occurs posttranslationally via the Sec61 translocon and requires ATP. We classified preproteins into three groups: 1) those that comprise ≤100 amino acids are strongly dependent on Sec62 for efficient translocation; 2) those in the size range of 120-160 amino acids use the SRP pathway, albeit inefficiently, and therefore rely on Sec62 for efficient translocation; and 3) those larger than 160 amino acids depend on the SRP pathway to preserve a transient translocation competence independent of Sec62. Thus, unlike in yeast, the Sec62-dependent translocation pathway in mammalian cells serves mainly as a fail-safe mechanism to ensure efficient secretion of small proteins and provides cells with an opportunity to regulate secretion of small proteins independent of the SRP pathway.
Lakkaraju AK, Thankappan R, Mary C et al., Mol Biol Cell. 2012 Jul;23(14):2712-22. doi: 10.1091/mbc.E12-03-0228. Epub 2012 May 30.
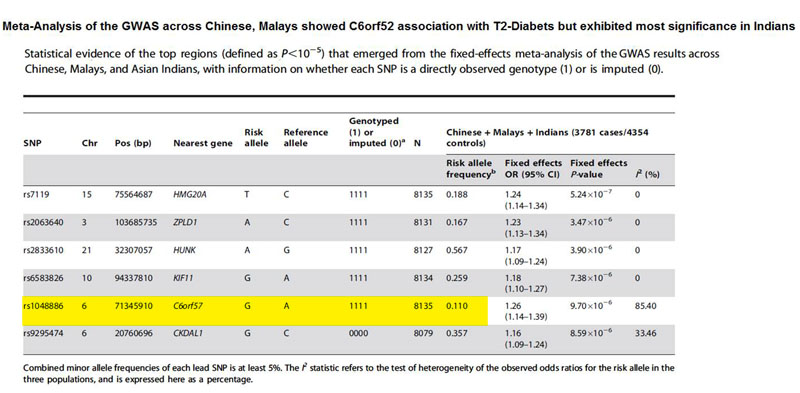
Recent large genome-wide association studies (GWAS) have identified multiple loci which harbor genetic variants associated with type 2 diabetes mellitus (T2D), many of which encode proteins not previously suspected to be involved in the pathogenesis of T2D. Most GWAS for T2D have focused on populations of European descent, and GWAS conducted in other populations with different ancestry offer a unique opportunity to study the genetic architecture of T2D. We performed genome-wide association scans for T2D in 3,955 Chinese (2,010 cases, 1,945 controls), 2,034 Malays (794 cases, 1,240 controls), and 2,146 Asian Indians (977 cases, 1,169 controls). In addition to the search for novel variants implicated in T2D, these multi-ethnic cohorts serve to assess the transferability and relevance of the previous findings from European descent populations in the three major ethnic populations of Asia, comprising half of the world's population. Of the SNPs associated with T2D in previous GWAS, only variants at CDKAL1 and HHEX/IDE/KIF11 showed the strongest association with T2D in the meta-analysis including all three ethnic groups. However, consistent direction of effect was observed for many of the other SNPs in our study and in those carried out in European populations. Close examination of the associations at both the CDKAL1 and HHEX/IDE/KIF11 loci provided some evidence of locus and allelic heterogeneity in relation to the associations with T2D. We also detected variation in linkage disequilibrium between populations for most of these loci that have been previously identified. These factors, combined with limited statistical power, may contribute to the failure to detect associations across populations of diverse ethnicity. These findings highlight the value of surveying across diverse racial/ethnic groups towards the fine-mapping efforts for the casual variants and also of the search for variants, which may be population-specific.
Sim X1, Ong RT, Suo C et al., PLoS Genet. 2011 Apr;7(4):e1001363. doi: 10.1371/journal.pgen.1001363. Epub 2011 Apr 7.
The human genome offers an attractive starting point for diabetes biomarker discovery. We have undertaken a survey of the Genetic Association Database (GAD) to develop a comprehensive genetic profiling of the type 1 and type 2 diabetes phenotypes. Using text mining, the GAD was explored for diabetes-associated genetic polymorphisms and a working database for type 1 and type 2 diabetes was established. In addition to well-characterized genes, 57 novel, uncharacterized Open Reading Frames (ORFs) encompassed in the dark matter of the human proteome were identified. Diverse bioinformatics and proteomics tools were used to characterize these ORFs for gene expression, protein motifs and domain information. Distinct protein classes including secreted products, enzymes, transporters, and receptors were encoded by these ORFs. Using expression Quantitative Traits Loci, Clinical Variations and the Genenome- Integrator tools, 50 novel ORFs associated with phenotypes for both type 1 and type 2 diabetes were identified. These results open up new avenues for better understanding type 1 and type 2 diabetes and may provide novel therapy targets for type 2 diabetes and associated disorders.
Delgado AP, Brandao P, Narayanan R, MOJ Proteomics Bioinform 1(4): (2014, July 19) 00020.
Xenin-25, a peptide co-secreted with the incretin hormone glucose-dependent insulinotropic polypeptide (GIP), possesses promising therapeutic actions for obesity-diabetes. However, native xenin-25 is rapidly degraded by serum enzymes to yield the truncated metabolites: xenin 9-25, xenin 11-25, xenin 14-25 and xenin 18-25. This study has examined the biological activities of these fragment peptides. In vitro studies using BRIN-BD11 cells demonstrated that native xenin-25 and xenin 18-25 possessed significant (P<0.05 to P<0.001) insulin-releasing actions at 5.6 and 16.7 mM glucose, respectively, but not at 1.1 mM glucase. In addition, xenin 18-25 significantly (P<0.05) potentiated the insulin-releasing action of the stable GIP mimetic (D-Ala2) GIP. In contrast, xenin 9-25, xenin 11-25 and xenin 14-25 displayed neither insulinotropic nor GIP-potentiating actions. Moreover,xenin 9-25, xenin 11-25 and xenin 14-25 significantly (P<0.05 to P<0.001) inhibited xenin-25 (10-8 M)-induced insulin release in vitro. I.p. administration of xenin-based peptides in combination with glucose to high fat-fed mice did not significantly affect the glycaemic excursion or glucose-induced insulin release compared with controls. However, when combined with (D-Ala2)GIP, all xenin peptides significantly (P<0.01 to P<0.001) reduced the overall glycaemic excursion, albeit to a similar extent as (D-Ala2)GIP alone. Xenin-25 and xenin 18-25 also imparted a potential synergistic effect on (D-Ala2) GIP-induced insulin release in high fat-fed mice. All xenin-based peptides lacked significant satiety effects in normal mice. These data demonstrate that the C-terminally derived fragment peptide of xenin-25, xenin 18-25, exhibits significant biological actions that could have therapeutic utility for obesity-diabetes.
Martin CM, Parthsarathy V, Pathak V, et al., J Endocrinol. 2014 Apr 22;221(2):193-200. doi: 10.1530/JOE-13-0617. Print 2014 May.
Xenin is a 25-amino acid peptide of the neurotensin/xenopsin family identified in gastric mucosa as well as in a number of tissues, including the pancreas of various mammals. In healthy subjects, plasma xenin immunoreactivity increases after meals. Infusion of the synthetic peptide in dogs evokes a rise in plasma insulin and glucagon levels and stimulates exocrine pancreatic secretion. The latter effect has also been demonstrated for xenin-8, the C-terminal octapeptide of xenin. We have investigated the effect of xenin-8 on insulin, glucagon and somatostatin secretion in the perfused rat pancreas. Xenin-8 stimulated basal insulin secretion and potentiated the insulin response to glucose in a dose-dependent manner (EC(50)=0.16 nM; R(2)=0.9955). Arginine-induced insulin release was also augmented by xenin-8 (by 40%; p<0.05). Xenin-8 potentiated the glucagon responses to both arginine (by 60%; p<0.05) and carbachol (by 50%; p<0.05) and counteracted the inhibition of glucagon release induced by increasing the glucose concentration. No effect of xenin-8 on somatostatin output was observed. Our observations indicate that the reported increases in plasma insulin and glucagon levels induced by xenin represent a direct influence of this peptide on the pancreatic B and A cells.
Silvestre et al. Regul Pept. 2003 Aug 15;115(1):25-9
Type I diabetes (T1D) is an autoimmune disease in which an immune response to pancreatic β-cells results in their loss over time. Although the conventional view is that this loss is due to autoimmune destruction, we present evidence of an additional phenomenon in which autoimmunity promotes islet endocrine cell transdifferentiation. The end result is a large excess of δ-cells, resulting from α- to β- to δ-cell transdifferentiation. Intermediates in the process of transdifferentiation were present in murine and human T1D. Here, we report that the peptide caerulein was sufficient in the context of severe β-cell deficiency to induce efficient induction of α- to β- to δ-cell transdifferentiation in a manner very similar to what occurred in T1D. This was demonstrated by genetic lineage tracing and time course analysis. Islet transdifferentiation proceeded in an islet autonomous manner, indicating the existence of a sensing mechanism that controls the transdifferentiation process within each islet. The finding of evidence for islet celltransdifferentiation in rodent and human T1D and its induction by a single peptide in a model of T1D has important implications for the development of β-cell regeneration therapies for diabetes.
Piran R, Lee SH, Li CR et al., Cell Death Dis. 2014 Jul 31;5:e1357. doi: 10.1038/cddis.2014.311.