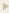1b. AICD Fragments derived from Prepro-APP Pathway
Schematic diagram of APP sequential processing (not drawn in
scale). EC: extracellular; TM: transmembrane; IC: intracellular. AB
domain is highlighted in red. For simplicity, only one cleavage site is
shown for each enzyme.
Hui Zheng and Edward H Koo, Mol Neurodegener.
2006; 1 : 5.
APP Intracellular Domain (AICD) Contributes to AD Pathology
Independently of AB
AICD (Short form of APP Intracellular Domain) (Human) / Beta-Amyloid Precursor (727-770), (018-69)
Long Form of APP Intracellular Domain (AICD) (Human) = Human Beta-Amyloid Precursor (712-770), (018-70)
APP Intracellular Domain (AICD) Related Products
%AICD%;%C-31%
1c. Other Soluble Fragments of Amyloid Precursor Proteins
Schematic of the different peptides administered to rats post-injury by Corrigan et. Al.
β-Amyloid Precursor Related Products
%018-65%;%018-66%;%018-67%
1a. Amyloid beta (1-42) and Fragments
Clinical and Biomarker Changes in Dominantly Inherited Alzheimerâs Disease.  Â
Clinical and Biomarker Changes in Dominantly Inherited Alzheimerâs Disease.  Â
Cross-Sectional Analyses of Clinical, Cognitive, Structural, Metabolic, and Biochemical Changes in Autosomal Dominant Alzheimerâs Disease Mutation Carriers versus Noncarriers, According to Estimated Years from Expected Symptom Onset. Dashed lines represent 95% confidence intervals of the fitted curves. SUVR denotes standardized uptake value ratio.
Truncated β-amyloid peptide channels provide an alternative mechanism for Alzheimer's Disease and Down syndrome.
Jang et al. Proc Natl Acad Sci U S A. 2010 Mar 22.
Truncated β-amyloid peptide channels provide an alternative mechanism for Alzheimer's Disease and Down syndrome.
Full-length amyloid beta peptides (Abeta(1-40/42)) form neuritic
amyloid plaques in Alzheimer's disease (AD) patients and are implicated
in AD pathology. However, recent transgenic animal models cast doubt on
their direct role in AD pathology. Nonamyloidogenic truncated
amyloid-beta fragments (Abeta(11-42) and Abeta(17-42)) are also found in
amyloid plaques of AD and in the preamyloid lesions of Down syndrome, a
model system for early-onset AD study. Very little is known about the
structure and activity of these smaller peptides, although they could be
the primary AD and Down syndrome pathological agents. Using
complementary techniques of molecular dynamics simulations, atomic force
microscopy, channel conductance measurements, calcium imaging, neuritic
degeneration, and cell death assays, we show that nonamyloidogenic
Abeta(9-42) and Abeta(17-42) peptides form ion channels with loosely
attached subunits and elicit single-channel conductances. The subunits
appear mobile, suggesting insertion of small oligomers, followed by
dynamic channel assembly and dissociation. These channels allow calcium
uptake in amyloid precursor protein-deficient cells. The channel
mediated calcium uptake induces neurite degeneration in human cortical
neurons. Channel conductance, calcium uptake, and neurite degeneration
are selectively inhibited by zinc, a blocker of amyloid ion channel
activity. Thus, truncated Abeta fragments could account for undefined
roles played by full length Abetas and provide a unique mechanism of AD
and Down syndrome pathologies. The toxicity of nonamyloidogenic peptides
via an ion channel mechanism necessitates a reevaluation of the current
therapeutic approaches targeting the nonamyloidogenic pathway as avenue
for AD treatment.
Jang et al. Proc Natl Acad Sci U S A. 2010 Mar 22.
Pyroglutamate-AB: Role in the natural history of Alzheimer's disease.
Gunn AP, et al. Int J Biochem Cell Biol. 2010 Dec;42(12):1915-8. Epub 2010 Sep 15.
Pyroglutamate-AB: Role in the natural history of Alzheimer's disease.
The accumulation of amyloid-beta (AB) peptides is believed to be a
central contributor to the neurodegeneration typically seen in
Alzheimer's disease (AD) brain. AB extracted from AD brains invariably
possesses extensive truncations, yielding peptides of differing N- and
C-terminal composition. Whilst AB is often abundant in the brains of
cognitively normal elderly people, the brains of AD patients are highly
enriched for N-terminally truncated AB bearing the pyroglutamate
modification. Pyroglutamate-AB (pE-AB) has a higher propensity for
oligomerisation and aggregation than full-length AB, potentially seeding
the accumulation of neurotoxic AB oligomers and amyloid deposits. In
addition, pE-AB has increased resistance to clearance by peptidases,
causing these peptides to persist in biological fluids and tissues. The
extensive deposition of pE-AB in human AD brain is under-represented in
many transgenic mouse models of AD, reflecting major differences in the
production and processing of AB peptides in these models compared to the
human disease state.
Gunn AP, et al. Int J Biochem Cell Biol. 2010 Dec;42(12):1915-8. Epub 2010 Sep 15.
Identification of low molecular weight pyroglutamate abeta oligomers in Alzheimer disease
Wirths O, et al. J Biol Chem. 2010 Dec 31;285(53):41517-24. Epub 2010 Oct 22.
Identification of low molecular weight pyroglutamate abeta oligomers in Alzheimer disease
A novel tool for therapy and diagnosis. N-terminally truncated AB
peptides starting with pyroglutamate (ABpE3) represent a major fraction
of all AB peptides in the brain of Alzheimer disease (AD) patients.
ABpE3 has a higher aggregation propensity and stability and shows
increased toxicity compared with full-length AB. In the present work, we
generated a novel monoclonal antibody (9D5) that selectively recognizes
oligomeric assemblies of ABpE3 and studied the potential involvement of
oligomeric ABpE3 in vivo using transgenic mouse models as well as human
brains from sporadic and familial AD cases. 9D5 showed an unusual
staining pattern with almost nondetectable plaques in sporadic AD
patients and non-demented controls. Interestingly, in sporadic and
familial AD cases prominent intraneuronal and blood vessel staining was
observed. Using a novel sandwich ELISA significantly decreased levels of
oligomers in plasma samples from patients with AD compared with healthy
controls were identified. Moreover, passive immunization of 5XFAD mice
with 9D5 significantly reduced overall AB plaque load and ABpE3 levels,
and normalized behavioral deficits. These data indicate that 9D5 is a
therapeutically and diagnostically effective monoclonal antibody
targeting low molecular weight ABpE3 oligomers.
Wirths O, et al. J Biol Chem. 2010 Dec 31;285(53):41517-24. Epub 2010 Oct 22.
Anti-11[E]-pyroglutamate-modified
amyloid B antibodies cross-react with other pathological AB species:
Relevance for immunotherapy.
Perez-Garmendia R, et al. J Neuroimmunol. 2010 Dec 15;229(1-2):248-55. Epub 2010 Sep 22.
Anti-11[E]-pyroglutamate-modified
amyloid B antibodies cross-react with other pathological AB species:
Relevance for immunotherapy.
N-truncated/modified forms of amyloid beta (AÃ) peptide are
found in diffused and dense core plaques in Alzheimer's disease (AD) and
Down's syndrome patients as well as animal models of AD, and represent
highly desirable therapeutic targets. In the present study we have
focused on N-truncated/modified AB peptide bearing amino-terminal
pyroglutamate at position 11 (ABN11(pE)). We identified two B-cell
epitopes recognized by rabbit anti-ABN11(pE) polyclonal antibodies.
Interestingly, rabbit anti-ABN11(pE) polyclonal antibodies bound also to
full-length AB1-42 and N-truncated/modified ABN3(pE), suggesting that
the three peptides may share a common B-cell epitope. Importantly,
rabbit anti-ABN11(pE) antibodies bound to naturally occurring AB
aggregates present in brain samples from AD patients. These results are
potentially important for developing novel immunogens for targeting
N-truncated/modified AB aggregates as well, since the most commonly used
immunogens in the majority of vaccine studies have been shown to induce
antibodies that recognize the N-terminal immunodominant epitope (EFRH)
of the full length AB, which is absent in N-amino truncated peptides.
Perez-Garmendia R, et al. J Neuroimmunol. 2010 Dec 15;229(1-2):248-55. Epub 2010 Sep 22.
Amyloid-β Related Products
%Amyloid beta%
1b. AICD Fragments derived from Prepro-APP Pathway
A mutation in APP protects against Alzheimer's disease and age-related cognitive decline.
A mutation in APP protects against Alzheimer's disease and age-related cognitive decline.
The prevalence of dementia in the Western world in people over the age of 60 has been estimated to be greater than 5%, about two-thirds of which are due to Alzheimer's disease. The age-specific prevalence of Alzheimer's disease nearly doubles every 5 years after age 65, leading to a prevalence of greater than 25% in those over the age of 90 (ref. 3). Here, to search for low-frequency variants in the amyloid-β precursor protein (APP) gene with a significant effect on the risk of Alzheimer's disease, we studied coding variants in APP in a set of whole-genome sequence data from 1,795 Icelanders. We found a coding mutation (A673T) in the APP gene that protects against Alzheimer's disease and cognitive decline in the elderly without Alzheimer's disease. This substitution is adjacent to the aspartyl protease β-site in APP, and results in an approximately 40% reduction in the formation of amyloidogenic peptides in vitro. The strong protective effect of the A673T substitution against Alzheimer's disease provides proof of principle for the hypothesis that reducing the β-cleavage of APP may protect against the disease. Furthermore, as the A673T allele also protects against cognitive decline in the elderly without Alzheimer's disease, the two may be mediated through the same or similar mechanisms.
(A) Brain lysates from 13 AD patients (AD) and 12 non-dementia
control subjects (Non-AD) were analyzed by SDS/PAGE and Western blotted
with 0443 antibody (top rows) that recognizes APP-C terminus or AT-8
antibody (bottom rows). Blots were stripped and reprobed with GAPDH
(middle rows). The upper rows show APP-CTF and AICD levels, which were
quantified by NIH ImageJ software (B). Although there was a large
variation in the AICD levels, they were significantly elevated in AD
patients compared to non-demented controls (P = 0.0017). The lane marked
with * in A (right panel) marks the non-AD sample with abnormally high
AICD levels (an outlier). The wide variation in AD samples is also seen
in phospho-tau levels. (C) Summary of pathological events observed in
FeCγ25 mice. AICD expression is driven by a CaMKIIα promoter, which
becomes active around P15, and activation of GSK-3B is observed at 6â8
weeks. Increased phosphorylation of tau is first observed at 3â4 months
of age, although tau does not become aggregated until 7â8 months of age.
Memory deficits become first apparent at this time point. Neuronal loss
and up-regulation of NPY are not observed up to 12â15 months of age but
become apparent >18 months of age. AD-related pathological features
described in this study are shown in open ovals.
Ghosal K., et al. PNAS, 2009,106 (43):18367-18372
Alzheimer's-related endosome dysfunction in Down syndrome is
Abeta-independent but requires APP and is reversed by BACE-1 inhibition.
Jiang, Y, et al. Proc Natl Acad Sci U S A. 2010 Jan 26;107(4):1630-5.
Alzheimer's-related endosome dysfunction in Down syndrome is
Abeta-independent but requires APP and is reversed by BACE-1 inhibition.
An additional copy of the beta-amyloid precursor protein (APP) gene
causes early-onset Alzheimer's disease (AD) in trisomy 21 (DS). Endosome
dysfunction develops very early in DS and AD and has been implicated in
the mechanism of neurodegeneration. Here, we show that morphological
and functional endocytic abnormalities in fibroblasts from individuals
with DS are reversed by lowering the expression of APP or
beta-APP-cleaving enzyme 1 (BACE-1) using short hairpin RNA constructs.
By contrast, endosomal pathology can be induced in normal disomic (2N)
fibroblasts by overexpressing APP or the C-terminal APP fragment
generated by BACE-1 (betaCTF), all of which elevate the levels of
betaCTFs. Expression of a mutant form of APP that cannot undergo
beta-cleavage had no effect on endosomes. Pharmacological inhibition of
APP gamma-secretase, which markedly reduced Abeta production but raised
betaCTF levels, also induced AD-like endosome dysfunction in 2N
fibroblasts and worsened this pathology in DS fibroblasts. These
findings strongly implicate APP and the betaCTF of APP, and exclude
Abeta and the alphaCTF, as the cause of endocytic pathway dysfunction in
DS and AD, underscoring the potential multifaceted value of BACE-1
inhibition in AD therapeutics.
Jiang, Y, et al. Proc Natl Acad Sci U S A. 2010 Jan 26;107(4):1630-5.
Mitochondrial {gamma}-secretase participates in the metabolism of mitochondria-associated amyloid precursor protein.
Mitochondrial {gamma}-secretase participates in the metabolism of mitochondria-associated amyloid precursor protein.
Intracellular amyloid-β peptide (Aβ) has been implicated in the pathogenesis of Alzheimer`s disease (AD). Mitochondria were found to be the target both for amyloid precursor protein (APP) that accumulates in the mitochondrial import channels and for Aβ that interacts with several proteins inside mitochondria and leads to mitochondrial dysfunction. Here, we have studied the role of mitochondrial γ-secretase in processing different substrates. We found that a significant proportion of APP is associated with mitochondria in cultured cells and that γ-secretase cleaves the shedded C-terminal part of APP identified as C83 associated with the outer membrane of mitochondria (OMM). Moreover, we have established the topology of the C83 in the OMM and found the APP intracellular domain (AICD) to be located inside mitochondria. Our data show for the first time that APP is a substrate for the mitochondrial γ-secretase and that AICD is produced inside mitochondria. Thus, we provide a mechanistic view of the mitochondria-associated APP metabolism where AICD, P3 peptide and potentially Aβ are produced locally and may contribute to mitochondrial dysfunction in AD.-Pavlov, P. F., Wiehager, B., Sakai, J., Frykman, S., Behbahani, H., Winblad, B., Ankarcrona, M. Mitochondrial γ-secretase participates in the metabolism of mitochondria-associated amyloid precursor protein.
Phosphorylation of APP-CTF-AICD domains and interaction with adaptor proteins: signal transduction and/or transcriptional role - relevance for Alzheimer pathology.
Phosphorylation of APP-CTF-AICD domains and interaction with adaptor proteins: signal transduction and/or transcriptional role - relevance for Alzheimer pathology.
In recent decades, the study of the amyloid precursor protein (APP) and of its proteolytic products carboxy terminal fragment (CTF), APP intracellular C-terminal domain (AICD) and amyloid beta has been mostly focussed on the role of APP as a producer of the toxic amyloid beta peptide. Here, we reconsider the role of APP suggesting, in a provocative way, the protein as a central player in a putative signalling pathway. We highlight the presence in the cytosolic tail of APP of the YENPTY motif which is typical of tyrosine kinase receptors, the phosphorylation of the tyrosine, serine and threonine residues, the kinases involved and the interaction with intracellular adaptor proteins. In particular, we examine the interaction with Shc and Grb2 regulators, which through the activation of Ras proteins elicit downstream signalling events such as the MAPK pathway. The review also addresses the interaction of APP, CTFs and AICD with other adaptor proteins and in particular with Fe65 for nuclear transcriptional activity and the importance of phosphorylation for sorting the secretases involved in the amyloidogenic or non-amyloidogenic pathways. We provide a novel perspective on Alzheimer's disease pathogenesis, focussing on the perturbation of the physiological activities of APP-CTFs and AICD as an alternative perspective from that which normally focuses on the accumulation of neurotoxic proteolytic fragments.
Alzheimer's disease-like pathological features in transgenic mice expressing the APP intracellular domain
Ghosal K., et al. PNAS, 2009,106 (43):18367-18372
Alzheimer's disease-like pathological features in transgenic mice expressing the APP intracellular domain
The hypothesis that amyloid-β (Aβ) peptides are the primary cause of Alzheimer's disease (AD) remains the best supported theory of AD pathogenesis. Yet, many observations are inconsistent with the hypothesis. Aβ peptides are generated when amyloid precursor protein (APP) is cleaved by presenilins, a process that also produces APP intracellular domain (AICD). We previously generated AICD-overexpressing transgenic mice that showed abnormal activation of GSK-3β, a pathological feature of AD. We now report that these mice exhibit additional AD-like characteristics, including hyperphosphorylation and aggregation of tau, neurodegeneration and working memory deficits that are prevented by treatment with lithium, a GSK-3β inhibitor. Consistent with its potential role in AD pathogenesis, we find AICD levels to be elevated in brains from AD patients. The in vivo findings that AICD can contribute to AD pathology independently of Aβ have important therapeutic implications and may explain some observations that are discordant with the amyloid hypothesis.
Ghosal K., et al. PNAS, 2009,106 (43):18367-18372
Αβ Hinders Nuclear Targeting of AICD and Fe65 in Primary Neuronal Cultures
A. G. Henriques et al. J Mol Neurosci. 2009 September; 39(1-2): 248–255.
Αβ Hinders Nuclear Targeting of AICD and Fe65 in Primary Neuronal Cultures
The intracellular domain of the Alzheimer’s amyloid precursor protein (AICD) has been described as an important player in the transactivation of specific genes. It results from proteolytic processing of the Alzheimer’s amyloid precursor protein (APP), as does the neurotoxic Aβ peptide. Although normally produced in cells, Aβ is typically considered to be a neurotoxic peptide, causing devastating effects. By exposing primary neuronal cultures to relatively low Aβ concentrations, this peptide was shown to affect APP processing. Our findings indicate that APP C-terminal fragments are increased with concomitant reduction in the expression levels of APP itself. AICD nuclear immunoreactivity detected under control conditions was dramatically reduced in response to Aβ exposure. Additionally, intracellular protein levels of Fe65 and GSK3 were also decreased in response to Aβ. APP nuclear signaling is altered by Aβ, affecting not only AICD production but also its nuclear translocation and complex formation with Fe65. In effect, Aβ can trigger a physiological negative feedback mechanism that modulates its own production.
A. G. Henriques et al. J Mol Neurosci. 2009 September; 39(1-2): 248–255.
APP Intracellular Domain (AICD) Related Products
%AICD%;%C-31%
1c. Other Soluble Fragments of Amyloid Precursor Proteins
Variant of TREM2 associated with the risk of Alzheimer's disease.
Variant of TREM2 associated with the risk of Alzheimer's disease.
BACKGROUND:
Sequence variants, including the ε4 allele of apolipoprotein E, have been associated with the risk of the common late-onset form of Alzheimer's disease. Few rare variants affecting the risk of late-onset Alzheimer's disease have been found.
METHODS:
We obtained the genome sequences of 2261 Icelanders and identified sequence variants that were likely to affect protein function. We imputed these variants into the genomes of patients with Alzheimer's disease and control participants and then tested for an association with Alzheimer's disease. We performed replication tests using case-control series from the United States, Norway, The Netherlands, and Germany. We also tested for a genetic association with cognitive function in a population of unaffected elderly persons.
RESULTS:
A rare missense mutation (rs75932628-T) in the gene encoding the triggering receptor expressed on myeloid cells 2 (TREM2), which was predicted to result in an R47H substitution, was found to confer a significant risk of Alzheimer's disease in Iceland (odds ratio, 2.92; 95% confidence interval [CI], 2.09 to 4.09; P=3.42Ã10(-10)). The mutation had a frequency of 0.46% in controls 85 years of age or older. We observed the association in additional sample sets (odds ratio, 2.90; 95% CI, 2.16 to 3.91; P=2.1Ã10(-12) in combined discovery and replication samples). We also found that carriers of rs75932628-T between the ages of 80 and 100 years without Alzheimer's disease had poorer cognitive function than noncarriers (P=0.003).
CONCLUSIONS:
Our findings strongly implicate variant TREM2 in the pathogenesis of Alzheimer's disease. Given the reported antiinflammatory role of TREM2 in the brain, the R47H substitution may lead to an increased predisposition to Alzheimer's disease through impaired containment of inflammatory processes. (Funded by the National Institute on Aging and others.).
The neuroprotective domains of the
amyloid precursor protein, in traumatic brain injury, are located in the
two growth factor domains.
Corrigan F, et al. Brain Res. 2011 Mar 10;1378:137-43. Epub 2011 Jan 6.
The neuroprotective domains of the
amyloid precursor protein, in traumatic brain injury, are located in the
two growth factor domains.
Abstract
The amyloid precursor protein (APP) is known to increase following
traumatic brain injury (TBI). This increase in levels of APP may be
deleterious to outcome due to the production of neurotoxic AB.
Conversely, this upregulation may be beneficial as cleavage of APP via
the alternative non-amyloidogenic pathway produces the soluble α form of
APP (sAPPα), which is known to have many neuroprotective and
neurotrophic functions. Indeed it has previously been shown that
treatment with sAPPα following a diffuse injury in rats improves
outcome. However, the exact location within the sAPPα molecule which
contains this neuroprotective activity has yet to be determined. The
sAPPα peptide can consist of up to 6 domains, with the main isoform in
the brain missing the 4th and 5th. Of the remaining domains, the D1 and
D6a domains seem the most likely as they have been shown to have
beneficial actions in vitro. This present study examined the effects of
in vivo posttraumatic administration via an intracerebroventricular
injection of the D1, D2 and D6a domains of sAPPα on outcome following
moderate-impact acceleration TBI in rats. While treatment with either
the D1 or D6a domains was found to significantly improve motor and
cognitive outcome, as assessed on the rotarod and Y maze, treatment with
the D2 domain had no effect. Furthermore axonal injury was reduced in
D1 and D6a domain treated animals, but not those that received the D2
domain. As the D1 and D6a domains contain a heparin binding region while
the D2 domain does not, this suggests that sAPPα mediates its
neuroprotective response through its ability to bind to heparin sulfate
proteoglycans.
Corrigan F, et al. Brain Res. 2011 Mar 10;1378:137-43. Epub 2011 Jan 6.
Amino acid sequence RERMS represents the active domain of amyloid beta/A4 protein precursor that promotes fibroblast growth.
Ninomiya H et al. J Cell Biol. 1993 May;121(4):879-86.
Amino acid sequence RERMS represents the active domain of amyloid beta/A4 protein precursor that promotes fibroblast growth.
Abstract
The growth of A-1 fibroblasts depends on exogenous amyloid beta/A4
protein precursor (APP), providing a simple bioassay to study the
function of APP. Our preliminary study, testing the activity of a series
of fragments derived from the secreted form of APP-695 (sAPP-695) on
this bioassay, has shown that at least one of the active sites of
sAPP-695 was localized within a 40-mer sequence (APP296-335, Kang
sequence; Roch, J.-M., I. P. Shapiro, M. P. Sundsmo, D. A. C. Otero, L.
M. Refolo, N. K. Robakis, and T. Saitoh. 1992. J. Biol. Chem.
267:2214-2221). In the present study, to further characterize the
growth-promoting activity of sAPP-695 on fibroblasts, we applied a
battery of synthetic peptides on this bioassay and found that: (a) the
sequence of five amino acids, RERMS (APP328-332, Phoenix Cat. # 018-09),
was uniquely required for the growth-promoting activity of sAPP-695;
(b) the activity was sequence-specific because the reverse-sequence
peptide of the active domain had no activity; and (c) the
four-amino-acid peptide RMSQ (APP330-333), which partially overlaps the
COOH-terminal side of the active sequence RERMS, could antagonize the
activity of sAPP-695. Furthermore, a recombinant protein which lacks
this active domain (APP20-591 without 306-335) did not promote
fibroblast cell growth, suggesting that this domain is the only site of
sAPP-695 involved in the growth stimulation. The availability of these
biologically active, short peptides and their antagonists should prove
to be an essential step for the elucidation ofAPPInvolvement in
regulation of cellular homeostasis.
Ninomiya H et al. J Cell Biol. 1993 May;121(4):879-86.
A novel neurotrophic peptide: APP63-73.
Wang R et al. Neuroreport. 2004 Dec 3;15(17):2677-80.
A novel neurotrophic peptide: APP63-73.
Abstract:
The objective of this study was to find out which N-terminal
segment/s of amyloid precursor protein (APP) has any neurotrophic
properties, since soluble APP-alpha (sAPP-alpha) has neurotrophic
effects. We investigated neurotrophic properties of eight peptide
segments of N-terminal APP. The APP63-73 was able to enhance neuronal
growth; augment axonal and cell body growth in human neuroblastoma cell
line, SH-SY5Y. Neurotrophic effects of chronic APP63-73 treatment were
assessed in vivo using streptozotocin-induced diabetes and
ovariectomized rats. Thus, this study demonstrated that APP63-73 peptide
has neurotrophic effects both in vivo and in vitro.
PMID: 15570177
Wang R et al. Neuroreport. 2004 Dec 3;15(17):2677-80.
Decrease in brain soluble amyloid precursor protein B (sAPPB) in Alzheimer's disease cortex.
Wu G et al. J Neurosci Res. 2011 Jun;89(6):822-32. doi: 10.1002/jnr.22618. Epub 2011 Mar 23.
Decrease in brain soluble amyloid precursor protein B (sAPPB) in Alzheimer's disease cortex.
Abstract
Amyloid-B peptide (AB) is generated by sequential cleavage of the
amyloid precursor protein (APP) by B-site amyloid precursor protein
cleaving enzyme 1 (B-secretase, or BACE1) and γ-secretase. Several
reports demonstrate increased BACE1 enzymatic activity in brain and
cerebrospinal fluid (CSF) from Alzheimer's disease (AD) subjects,
suggesting that an increase in BACE1-mediated cleavage of APP drives
amyloid pathophysiology in AD. BACE1 cleavage of APP leads to the
generation of a secreted N-terminal fragment of APP (sAPPB). To relate
BACE1 activity better to endogenous APP processing in AD and control
brains, we have directly measured brain sAPPB levels using a novel APP
B-site specific enzyme-linked immunosorbent assay. We demonstrate a
significant reduction in brain cortical sAPPB levels in AD compared with
control subjects. In the same brain samples, BACE1 activity was
unchanged, full-length APP and sAPPα levels were significantly reduced,
and AB peptides were significantly elevated. In conclusion, a reduction
in cortical brain sAPPB together with unchanged BACE1 activity suggests
that this is due to reduced full-length APP substrate in late-stage AD
subjects. These results highlight the need for multiparameter analysis
of the amyloidogenic process to understand better AD pathophysiology in
early vs. late-stage AD.
Wu G et al. J Neurosci Res. 2011 Jun;89(6):822-32. doi: 10.1002/jnr.22618. Epub 2011 Mar 23.