|
|
|
Pyroglutamate Amyloid-β Peptides |

|
BACKGROUND: The amyloid-β (Aβ) peptide is the primary component of the extracellular senile plaques characteristic of Alzheimer's disease (AD). The metals hypothesis implicates redox-active copper ions in the pathogenesis of AD and the Cu(2+) coordination of various Aβ peptides has been widely studied. A number of disease-associated modifications involving the first 3 residues are known, including isomerisation, mutation, truncation and cyclisation, but are yet to be characterised in detail. In particular, Aβ in plaques contain a significant amount of truncated pyroglutamate species, which appear to correlate with disease progression. METHODOLOGY/PRINCIPAL FINDINGS: We previously characterised three Cu(2+)/Aβ1-16 coordination modes in the physiological pH range that involve the first two residues. Based upon our finding that the carbonyl of Ala2 is a Cu(2+) ligand, here we speculate on a hypothetical Cu(2+)-mediated intramolecular cleavage mechanism as a source of truncations beginning at residue 3. Using EPR spectroscopy and site-specific isotopic labelling, we have also examined four Aβ peptides with biologically relevant N-terminal modifications, Aβ1[isoAsp]-16, Aβ1-16(A2V), Aβ3-16 and Aβ3[pE]-16. The recessive A2V mutation preserved the first coordination sphere of Cu(2+)/Aβ, but altered the outer coordination sphere. Isomerisation of Asp1 produced a single dominant species involving a stable 5-membered Cu(2+) chelate at the amino terminus. The Aβ3-16 and Aβ3[pE]-16 peptides both exhibited an equilibrium between two Cu(2+) coordination modes between pH 6-9 with nominally the same first coordination sphere, but with a dramatically different pH dependence arising from differences in H-bonding interactions at the N-terminus. CONCLUSIONS/SIGNIFICANCE: N-terminal modifications significantly influence the Cu(2+) coordination of Aβ, which may be critical for alterations in aggregation propensity, redox-activity, resistance to degradation and the generation of the Aβ3-× (× = 40/42) precursor of disease-associated Aβ3[pE]-x species.

Drew SC, et al. PLoS One. 2010 Dec 30;5(12):e15875.
|
|
|
|
Glutaminyl cyclase (QC) was discovered recently as the enzyme catalyzing the pyroglutamate (pGlu or pE) modification of N-terminally truncated Alzheimer's disease (AD) Abeta peptides in vivo. This modification confers resistance to proteolysis, rapid aggregation and neurotoxicity and can be prevented by QC inhibitors in vitro and in vivo, as shown in transgenic animal models. However, in mouse brain QC is only expressed by a relatively low proportion of neurons in most neocortical and hippocampal subregions. Here, we demonstrate that QC is highly abundant in subcortical brain nuclei severely affected in AD. In particular, QC is expressed by virtually all urocortin-1-positive, but not by cholinergic neurons of the Edinger-Westphal nucleus, by noradrenergic locus coeruleus and by cholinergic nucleus basalis magnocellularis neurons in mouse brain. In human brain, QC is expressed by both, urocortin-1 and cholinergic Edinger-Westphal neurons and by locus coeruleus and nucleus basalis Meynert neurons. In brains from AD patients, these neuronal populations displayed intraneuronal pE-Abeta immunoreactivity and morphological signs of degeneration as well as extracellular pE-Abeta deposits. Adjacent AD brain structures lacking QC expression and brains from control subjects were devoid of such aggregates. This is the first demonstration of QC expression and pE-Abeta formation in subcortical brain regions affected in AD. Our results may explain the high vulnerability of defined subcortical neuronal populations and their central target areas in AD as a consequence of QC expression and pE-Abeta formation.
Morawski M, et al. Acta Neuropathol. 2010 Aug;120(2):195-207. Epub 2010 Apr 10.
|
|

|
Morawski M et al., Acta Neuropathol. 2010 Aug;120(2):195 207
|
|
|

In the EWN of control subjects there was only a faint intraneuronal pE-Aβ immunoreactivity. In contrast, in brains
from AD patients the EWN was robustly labelled with intraneuronal pE-Aβ (arrows) and pE-Aβ plaques (asterisks).
Morawski M et al., Acta Neuropathol. 2010 Aug;120(2):195 207
|
|
|
|
A number of distinct beta-amyloid (Abeta) variants or multimers have been implicated in Alzheimer's disease (AD), and antibodies recognizing such peptides are in clinical trials. Humans have natural Abeta-specific antibodies, but their diversity, abundance, and function in the general population remain largely unknown. Here, we demonstrate with peptide microarrays the presence of natural antibodies against known toxic Abeta and amyloidogenic non-Abeta species in plasma samples and cerebrospinal fluid of AD patients and healthy controls aged 21-89 years. Antibody reactivity was most prominent against oligomeric assemblies of Abeta and pyroglutamate or oxidized residues, and IgGs specific for oligomeric preparations of Abeta1-42 in particular declined with age and advancing AD. Most individuals showed unexpected antibody reactivities against peptides unique to autosomal dominant forms of dementia (mutant Abeta, ABri, ADan) and IgGs isolated from plasma of AD patients or healthy controls protected primary neurons from Abeta toxicity. Aged vervets showed similar patterns of plasma IgG antibodies against amyloid peptides, and after immunization with Abeta the monkeys developed high titers not only against Abeta peptides but also against ABri and ADan peptides. Our findings support the concept of conformation-specific, cross-reactive antibodies that may protect against amyloidogenic toxic peptides. If a therapeutic benefit of Abeta antibodies can be confirmed in AD patients, stimulating the production of such neuroprotective antibodies or passively administering them to the elderly population may provide a preventive measure toward AD.
Britschgi M, et al. Proc Natl Acad Sci U S A. 2009 Jul 21;106(29):12145-50. Epub 2009 Jul 6
|
|
|
|
Biritschgi M, et al. PNAS. 2009 Jul 21;106(29):12145-50
|
|
|
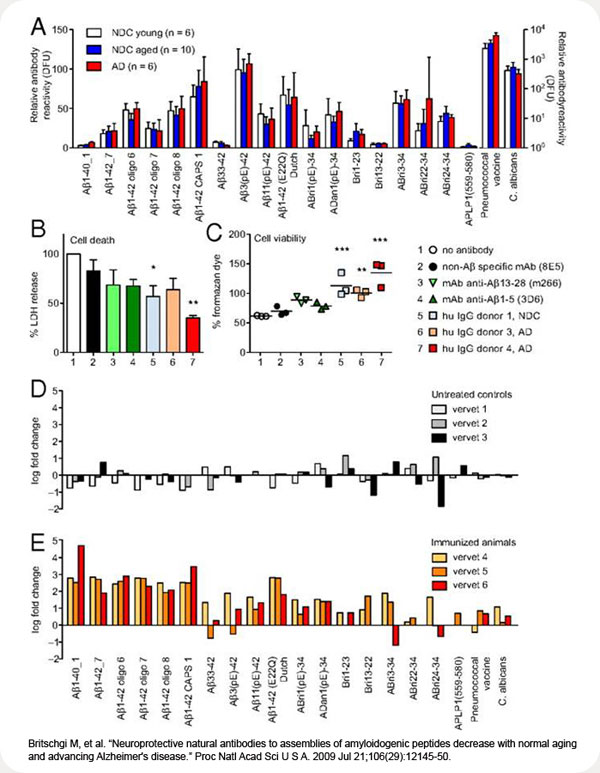
(A) IgG reactivity pattern in CSF of AD patients (≥70 years) and NDC young (24–48 years) and aged individuals (58–83 years). Bars represent means of median reactivities ± SEM. (B and C) Protective effect of human IgGs on primary mouse E16 neurons incubated with oligomeric preparation of Aβ1-42 was measured by percentage LDH release in dying cells (B) and percentage generation of formazan (C). Amount of water-soluble formazan produced by surviving cells in culture medium alone was considered 100% survival. Bars represent mean ± SEM of 3 experiments done in triplicates. Symbols in scatter plot represent mean of 1 triplicate experiment. *, P ≤ 0.05, 1-way ANOVA and Dunnett's multiple comparison test with “no antibody” as control. (D and E) Vervet monkeys were left untreated (D) or immunized with a mixture of Aβ1-40 and Aβ1-42 (E) (39). Immunoreactivity at the end of the study (day 301) was divided by the baseline reactivity to individual peptides, and the result is presented as log fold change. Bars represent median fold change of reactivity to a peptide for each individual animal. As expected, immunoreactivity to Aβ1-40 and Aβ1-42 peptides and oligomeric assemblies increase over several log ranges. Note the increase in reactivity to Aβ11(pE)-42, ABri, and ADan but the lack of increase to Aβ33–42. DFU, digital fluorescent units.
Biritschgi M, et al. PNAS. 2009 Jul 21;106(29):12145-50
|
|
|
The accumulation of amyloid-beta (Aβ) peptides is believed to be a central contributor to the neurodegeneration typically seen in Alzheimer's disease (AD) brain. Aβ extracted from AD brains invariably possesses extensive truncations, yielding peptides of differing N- and C-terminal composition. Whilst Aβ is often abundant in the brains of cognitively normal elderly people, the brains of AD patients are highly enriched for N-terminally truncated Aβ bearing the pyroglutamate modification. Pyroglutamate-Aβ (pE-Aβ) has a higher propensity for oligomerisation and aggregation than full-length Aβ, potentially seeding the accumulation of neurotoxic Aβ oligomers and amyloid deposits. In addition, pE-Aβ has increased resistance to clearance by peptidases, causing these peptides to persist in biological fluids and tissues. The extensive deposition of pE-Aβ in human AD brain is under-represented in many transgenic mouse models of AD, reflecting major differences in the production and processing of Aβ peptides in these models compared to the human disease state.
Gunn AP, et al. Int J Biochem Cell Biol. 2010 Dec;42(12):1915-8. Epub 2010 Sep 15.
|
|
|
N-terminally truncated Aβ peptides starting with pyroglutamate (AβpE3) represent a major fraction of all Aβ peptides in the brain of Alzheimer disease (AD) patients. AβpE3 has a higher aggregation propensity and stability and shows increased toxicity compared with full-length Aβ. In the present work, we generated a novel monoclonal antibody (9D5) that selectively recognizes oligomeric assemblies of AβpE3 and studied the potential involvement of oligomeric AβpE3 in vivo using transgenic mouse models as well as human brains from sporadic and familial AD cases. 9D5 showed an unusual staining pattern with almost nondetectable plaques in sporadic AD patients and non-demented controls. Interestingly, in sporadic and familial AD cases prominent intraneuronal and blood vessel staining was observed. Using a novel sandwich ELISA significantly decreased levels of oligomers in plasma samples from patients with AD compared with healthy controls were identified. Moreover, passive immunization of 5XFAD mice with 9D5 significantly reduced overall Aβ plaque load and AβpE3 levels, and normalized behavioral deficits. These data indicate that 9D5 is a therapeutically and diagnostically effective monoclonal antibody targeting low molecular weight AβpE3 oligomers.
Wirths O, et al. J Biol Chem. 2010 Dec 31;285(53):41517-24. Epub 2010 Oct 22.
|
|
|
|
N-truncated/modified forms of amyloid beta (Aß) peptide are found in diffused and dense core plaques in Alzheimer's disease (AD) and Down's syndrome patients as well as animal models of AD, and represent highly desirable therapeutic targets. In the present study we have focused on N-truncated/modified Aβ peptide bearing amino-terminal pyroglutamate at position 11 (AβN11(pE)). We identified two B-cell epitopes recognized by rabbit anti-AβN11(pE) polyclonal antibodies. Interestingly, rabbit anti-AβN11(pE) polyclonal antibodies bound also to full-length Aβ1-42 and N-truncated/modified AβN3(pE), suggesting that the three peptides may share a common B-cell epitope. Importantly, rabbit anti-AβN11(pE) antibodies bound to naturally occurring Aβ aggregates present in brain samples from AD patients. These results are potentially important for developing novel immunogens for targeting N-truncated/modified Aβ aggregates as well, since the most commonly used immunogens in the majority of vaccine studies have been shown to induce antibodies that recognize the N-terminal immunodominant epitope (EFRH) of the full length Aβ, which is absent in N-amino truncated peptides
Perez-Garmendia R, et al. J Neuroimmunol. 2010 Dec 15;229(1-2):248-55. Epub 2010 Sep 22.
|
|
|
|
A proposed key event in the pathogenesis of Alzheimer's disease (AD) is the formation of neurotoxic amyloid beta (Abeta) oligomers and amyloid plaques in specific brain regions that are affected by the disease. The main plaque component is the 42 amino acid isoform of Alphabeta (Abeta1-42), which is thought to initiate plaque formation and AD pathogenesis. Numerous isoforms of Abeta, e.g., Abeta1-42, Abeta1-40 and the 3-pyroglutamate derivate of Abeta3-42 (pGluAbeta3-42), have been detected in the brains of sporadic AD (SAD) and familial AD (FAD) subjects. However, the relative importance of these isoforms in the pathogenesis of AD is not fully understood. Here, we report a detailed study using immunoprecipitation in combination with mass spectrometric analysis to determine the Abeta isoform pattern in the cerebellum, cortex and hippocampus in AD, including subjects with a mutation in the presenilin (M146V) or amyloid precursor protein (KM670/671NL) genes, SAD subjects and non-demented controls. We show that the dominating Abeta isoforms in the three different brain regions analyzed from control, SAD, and FAD are Abeta1-42, pGluAbeta3-42, Abeta4-42 and Abeta1-40 of which Abeta1-42 and Abeta4-42 are the dominant isoforms in the hippocampus and the cortex in all groups analyzed, controls included. No prominent differences in Abeta isoform patterns between FAD and SAD patients were seen, underscoring the similarity in the amyloid pathology of these two disease entities.
Portelius E, et al. Acta Neuropathol. 2010 Aug;120(2):185-93
|
|
|
|
|
amyloid_beta_peptides;Amyloid_beta_precursor_like
%018-63%;%018-64%
|
|
|