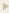|
|
|
AMPK (AMP-activated Protein Kinase) |
|
An Intracellular Energy Sensor Maintaining the Energy Balance. |
AMPK(AMP-activated Protein Kinase) an intracellular energy sensor maintaining the energy balance.AMP-activated protein kinase (AMPK) is the downstream component of a protein kinase cascade that acts as an intracellular energy sensor maintaining the energy balance within the cell. The finding that leptin and adiponectin activate AMPK to alter metabolic pathways in muscle and liver provides direct evidence for this role in peripheral tissues. The hypothalamus is a key regulator of food intake and energy balance, coordinating body adiposity and nutritional state in response to peripheral hormones, such as leptin, peptide YY(3-36) (PYY) and ghrelin. To date the hormonal regulation of AMPK in the hypothalamus, or its potential role in the control of food intake, have not been reported. Here we demonstrate that counter-regulatory hormones involved in appetite control regulate AMPK activity, and that pharmacological activation of AMPK in the hypothalamus increases food intake. In vivo administration of leptin, which leads to a reduction in food intake, decreases hypothalamic AMPK activity. By contrast, injection of ghrelin in vivo, which increases food intake, stimulates AMPK activity in the hypothalamus. Consistent with the effect of ghrelin, injection of 5-amino-4-imidazole carboxamide (AICA) riboside, a pharmacological activator of AMPK, into either the third cerebral ventricle or directly into the paraventricular nucleus of the hypothalamus significantly increased food intake. These results suggest that AMPK is regulated in the hypothalamus by hormones which regulate food intake. Furthermore, direct pharmacological activation of AMPK in the hypothalamus is sufficient to increase food intake. These findings demonstrate that AMPK plays a role in the regulation of feeding and identify AMPK as a novel target for anti-obesity drugs.

Human gene mutations affecting cardiac energetics and metabolism. Energy substrate utilization is directed by critical metabolic sensors in myocytes, including AMP-activated protein kinase (AMPK), which, in response to increased AMP/ATP levels, phosphorylates target proteins and thereby regulates glycogen and fatty acid metabolism, critical energy sources for the heart. Glycogen metabolism involves a large number of proteins including -galactosidase A (mutated in Fabry disease) and LAMP2 (mutated in Danon disease). Glycogen and fatty acids are substrates for multiprotein complexes located within the mitochondria for the synthesis of ATP. KATP channels composed of an enzyme complex and a potassium pore participate in decoding metabolic signals to maximize cellular functions during stress adaptation. Human mutations (orange text) that cause cardiomyopathies have been identified in the regulatory SUR2A subunit of KATP, the 2 subunit of AMPK, mitochondrial proteins, -galactosidase A, and LAMP2.
Morita H., et al. J. Clin. Invest. 115:518-526 (2005). doi:10.1172/JCI200524351.
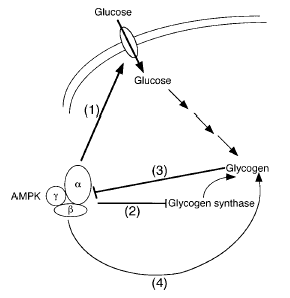
Role of AMPK in regulating energy balance
at the whole-body level. Green arrows indicate positive effects,
and red lines with bars indicate negative effects. In the
hypothalamus, activation of AMPK in response to low glucose
or leptin levels increases food intake (18, 19); references
for other effects of AMPK activation can be found in recent
reviews (1). FA, fatty acid.

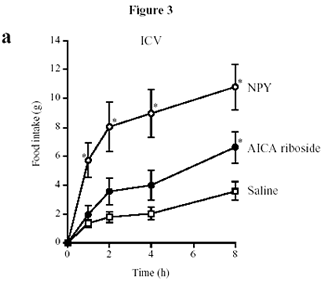
Role of AMPK in regulating energy balance
at the single-cell level. The way in which the AMPK system
controls the balance between ATP consumption (e.g., by biosynthesis,
cell growth, or muscle contraction) and ATP production via
catabolism is illustrated. If the rate of ATP consumption
exceeds its rate of production, ADP will tend to rise and
be converted to AMP by the enzyme adenylate kinase. The rise
in level of the activating ligand AMP, coupled with the fall
in level of the inhibitory nucleotide ATP, activates AMPK,
which then switches off ATP-consuming processes and switches
on catabolism in an attempt to redress the balance.
D. Grahame Hardie. J. Clin. Invest.
114:465-468 (2004). doi:10.1172/JCI200422683.

Proposed model for role of AMPK in anorexigenic
signalling in the hypothalamus. Anorexigenic signals activate
POMC neurons in ARH (arcuate hypothalamus) via STAT3 and possibly
also PI3 kinase, generating a second anorexigenic signal mediated
by -melanocyte stimulating hormone (-MSH). In contrast, the
anorexigenic signals suppress the activity of NPY/AGRP neurons,
partly via STAT3 and possibly also PI3 kinase, and decrease
AMPK activity in these neurons. Decreased AMPK activity enhances
the suppression of NPY/AGRP effects, leading to activation
of MC4 receptor signalling in PVH (paraventricular hypothalamus)
neurons. MC4 receptor activation decreases AMPK activity in
PVH, which probably further enhances neurotransmission required
for regulation of food intake and energy balance. Decreased
NPY signalling in PVH functionally enhances the MC4 receptor
signalling pathway. In addition, decreased AMPK activity in
other hypothalamic regions may enhance the MC4 receptor signalling
pathway by projections to the ARH or PVH (for example, NPY
neurons in DMH) and recruit additional pathways that may regulate
food intake.
MINOKOSHI Y., et al. Nature, 428, 569 - 574 (01 April 2004); doi:10.1038/nature02440
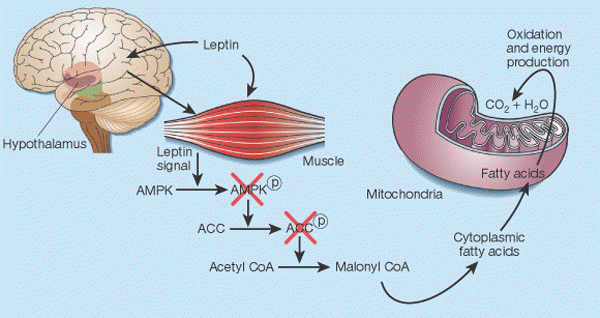
Leptin's control of fat in skeletal muscle2. In cells, there is a balance between transport of fatty acids into mitochondria and their subsequent oxidation, and storage of these compounds as triglycerides in the cytoplasm. This balance is regulated mainly by malonyl CoA, a fatty acid that is generated by the enzyme acetyl CoA carboxylase (ACC). Malonyl CoA inhibits transport of fatty acids into mitochondria, thereby preventing their oxidation12. Leptin causes the phosphorylation of AMP-activated protein kinase (AMPK), which in turn phosphorylates ACC, inactivating it13. Leptin thus inhibits malonyl CoA synthesis, leading to greater mitochondrial import and consumption of fatty acids. These events seem to result both from the direct action of leptin on skeletal muscle and from its indirect influence that operates through the hypothalamus.
Friedman J. Nature. 2002 Jan 17;415(6869):268-9

Figure 2 Model for the involvement of AMPK in the regulation of skeletal muscle glucose transport in response to AICAR, hypoxia, electrical stimulation and exercise. It is proposed that AICAR- and hypoxia-induced glucose uptake in skeletal muscle are AMPK-dependent, whereas exercise-induced glucose uptake is not or is only partially dependent on AMPK. It should be considered that during electrically stimulated muscle contraction (and perhaps exercise) a part of the stimulus to glucose transport may be due to hypoxia involving AMPK. Arrow thickness reflects the proposed relative involvement of the different pathways.
Nielsen JN, et al. Biochem Soc Trans. 2003 Feb;31(Pt 1):186-90
|
|
 |
| 
|
 |
| Malonyl-CoA
content of muscle is controlled by the relative
rate of synthesis by acetyl-CoA carboxylase (ACC)
and the rate of degradation by malonyl-CoA decarboxylase
(MCD). AMPK phosphorylates (P) and inactivates ACC.
On the basis of effects of 5-aminoimidazole- 4-carboxamide
-riboside (AICAR) on activation of MCD in incubated
extensor digitorum longus, AMPK is hypothesized
to phosphorylate and activate MCD (Ref. 80). These
changes could result in a decline in malonyl-CoA
(rat muscle studies).
Winder WW. J Appl Physiol. 2001 Sep;91(3):1017-28 |
Energy homeostasis and feeding are regulated by the central
nervous system (CNS). C75, a fatty acid synthase (FAS) inhibitor,
causes weight loss and anorexia, implying a novel CNS pathway(s)
for sensing energy balance. AMP-activated protein kinase (AMPK),
a sensor of peripheral energy balance, is phosphorylated and
activated when energy sources are low. Here, we identify a
role for hypothalamic AMPK in the regulation of feeding behavior
and in mediating C75’s anorexic effects. AICAR, an activator
of AMPK, increased food intake, whereas compound C, an inhibitor
of AMPK, decreased food intake. C75 rapidly reduced the level
of the phosphorylated AMPK a subunit (pAMPKα)
in the hypothalamus, even in fasted mice that had elevated
hypothalamic pAMPKa levels. Furthermore, AICAR reversed both
the C75-induced anorexia and the decrease in hypothalamic
pAMPKα levels. C75 elevated hypothalamic neuronal ATP levels, which
may contribute to the mechanism by which C75 decreased AMPK
activity. C75 reduced the levels of pAMPKα and phosphorylated cAMP response element binding protein (pCREB)
in the arcuate nucleus neurons of the hypothalamus, suggesting
a mechanism for the reduction in NPY expression seen with
C75 treatment. These data indicate that modulation of FAS
activity in the hypothalamus can alter energy perception via
AMPK, which functions as a physiological energy sensor in
the hypothalamus.
J. Biol. Chem, 10.1074/jbc.M402165200
AMP-activated protein kinase (AMPK) is the downstream component
of a protein kinase cascade that acts as an intracellular
energy sensor maintaining the energy balance within the cell.
The finding that leptin and adiponectin activate AMPK to alter
metabolic pathways in muscle and liver provides direct evidence
for this role in peripheral tissues. The hypothalamus is a
key regulator of food intake and energy balance, coordinating
body adiposity and nutritional state in response to peripheral
hormones, such as leptin, peptide YY(3-36) (PYY) and ghrelin.
To date the hormonal regulation of AMPK in the hypothalamus,
or its potential role in the control of food intake, have
not been reported. Here we demonstrate that counter-regulatory
hormones involved in appetite control regulate AMPK activity,
and that pharmacological activation of AMPK in the hypothalamus
increases food intake. In vivo administration of leptin, which
leads to a reduction in food intake, decreases hypothalamic
AMPK activity. By contrast, injection of ghrelin in vivo,
which increases food intake, stimulates AMPK activity in the
hypothalamus. Consistent with the effect of ghrelin, injection
of 5-amino-4-imidazole carboxamide (AICA) riboside, a pharmacological
activator of AMPK, into either the third cerebral ventricle
or directly into the paraventricular nucleus of the hypothalamus
significantly increased food intake. These results suggest
that AMPK is regulated in the hypothalamus by hormones which
regulate food intake. Furthermore, direct pharmacological
activation of AMPK in the hypothalamus is sufficient to increase
food intake. These findings demonstrate that AMPK plays a
role in the regulation of feeding and identify AMPK as a novel
target for anti-obesity drugs.
 |  |
 |
 |
Andersson U, Filipsson K, Abbott CR,
Woods A, Smith K, Bloom SR, Carling D, Small CJ. J Biol Chem.
2004 Jan 23 [Epub ahead of print]
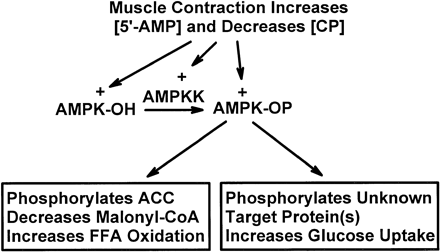
Postulated mechanisms of increase in fatty acid oxidation and on glucose uptake in skeletal muscle in response to contraction. AMPKK, AMP-activated protein kinase kinase; [5'-AMP] and [CP], 5'-AMP and creatine phosphate concentrations, respectively; AMPK-OH and AMPK-OP, nonphosphorylated and phosphorylated AMPK, respectively; ACC, acetyl-CoA carboxylase; FFA, free fatty acids.

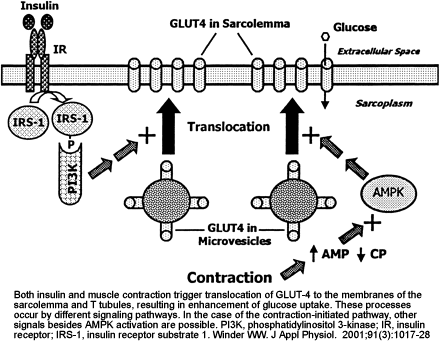
Two mechanisms for stimulation of glucose uptake in skeletal muscle, one mediated by insulin and one triggered by muscle contraction. The hypothesis of mediation of the contraction effect by AMPK is based on the observations that exercise and electrical stimulation increase AMPK activity and glucose uptake and that glucose uptake is increased by chemical activation of AMPK with AICA-riboside. IR, insulin receptor; IRS-1, insulin receptor substrate 1; PI 3-kinase, phosphatidylinositol 3-kinase.
Winder WW, Hardie DG. Am J Physiol. 1999 Jul;277(1 Pt 1):E1-10
Carling D. Trends Biochem Sci. 2004
Jan;29(1):18-24
Leclerc I, et
al. Am J Physiol Endocrinol Metab. 2004 Feb 10 [Epub ahead
of print]
Kim EK et al. JBC Papers in Press. Published on
March 17, 2004 as Manuscript M402165200.
|

|
Short-term
signals regulating food intake. Signals from the
GI tract and the liver are involved in short-term
regulation of feeding. Afferent signals travel in
vagal nerve fibers from stretch receptors, and chemoreceptors
activated by the presence of nutrients in the stomach
and proximal small intestine are involved in meal
termination. Nutrients arriving via the portal vein
may also trigger vagal afferent signals from the
liver. Glucose can modulate food intake by acting
on glucose-responsive neurons in the CNS. Ketones
appear to decrease appetite. In response to nutrient
stimulation, the proximal intestine releases cholecystokinin
(CCK), which reaches the liver via the portal vein
and the CNS via the systemic circulation; CCK may
act on CCK-A receptors at both sites to inhibit
food intake. Endocrine L cells in the terminal small
intestine (ileum) release glucagon-like peptide-1
(GLP-1), which inhibits feeding, most likely at
a hepatic site or by inhibiting gastric emptying.
The short-term signals by themselves do not produce
sustained alterations in energy intake and body
adiposity.
|
Long-term
signals regulating food intake and energy homeostasis.
Insulin and leptin are the two most important long-term
regulators of food intake and energy balance. Both
insulin and leptin act in the CNS to inhibit food
intake and to increase energy expenditure, most
likely by activating the sympathetic nervous system
(SNS). Insulin is secreted from ?cells in the endocrine
pancreas in response to circulating nutrients (glucose
and amino acids) and to the incretin hormones, glucose-dependent
insulinotropic polypeptide (GIP) and GLP-1, which
are released during meal ingestion and absorption.
Insulin can also act indirectly by stimulating leptin
production from adipose tissue via increased glucose
metabolism. In contrast, dietary fat and fructose
do not stimulate insulin secretion and therefore
do not increase leptin production. There is also
evidence that leptin can inhibit insulin secretion
from the pancreas. The gastric hormone ghrelin increases
food intake and decreases fat oxidation in rodents
and may have an anabolic role in long-term food
intake regulation. The long-term signals interact
with the short-term signals in the regulation of
energy homeostasis and appear to set sensitivity
to the satiety-producing effects of short-term signal
such as CCK.
Havel PJ.
Exp Biol Med (Maywood). 2001 Dec;226(11):963-77
|  |
|
|
|
003-98;077-66;077-66A
|
|
|